O que é o retinoblastoma?
O retinoblastoma, chamado popularmente de câncer ocular ou câncer nos olhos infantil, é um tumor maligno que surge na retina, uma fina camada composta por células nervosas que reveste o interior dos olhos e é responsável pela captação de luz. A retina “sente” a presença de luz e envia impulsos através do nervo óptico para o cérebro, onde esses sinais são interpretados como imagens.
O retinoblastoma é o tumor ocular mais comum da infância, sendo responsável por até 15% de todos os cânceres que surgem no primeiro ano de vida.
Ainda que possa surgir nos dois olhos, em mais de 75% dos casos, o tumor surge em apenas um deles.
O retinoblastoma ocorre em aproximadamente 1 a cada 15.000 crianças. A idade média no diagnóstico é de 18 a 20 meses; média de 12 meses para crianças com doença bilateral e 24 meses para crianças com doença unilateral. Em 95% dos casos, o tumor surge antes dos cinco anos de idade.
A incidência é semelhante em meninos e meninas, e não há predileção racial.
Se não for tratado a tempo, o retinoblastoma é uma doença mortal. Felizmente, com os avanços no tratamento, a sobrevida atualmente é de mais de 95%.
O encaminhamento precoce a um oftalmologista oncológico e o tratamento adequado por uma equipe multidisciplinar são essenciais para otimizar o resultado visual e a sobrevida da criança.
Como surge
O retinoblastoma é uma doença de origem genética, decorrente da mutação de um gene, chamado RB1, localizado no braço longo do cromossomo 13.
Essas mutações fazem com que as células da retina continuem crescendo e se multiplicando de forma descontrolada, enquanto as células saudáveis morrem. Essa massa acumulada de células forma o tumor dentro dos olhos.
As células de retinoblastoma podem invadir mais profundamente e alcançar estruturas ao redor dos olhos. Também pode se espalhar (causar metástases) para outras áreas do corpo, incluindo o cérebro e a coluna vertebral.
Forma hereditária
Em 30% dos casos, a doença tem origem hereditária, ou seja, a mutação do cromossomo 13 é herdada de um dos pais. Se um dos pais for portador de um gene mutante (gene RB1), cada filho ou filha terá 50% de chance de herdar esse gene.
Crianças com a forma hereditária de retinoblastoma tendem a desenvolver a doença mais cedo. O retinoblastoma hereditário também tende a ocorrer em ambos os olhos, ao contrário da forma não hereditária, que costuma provocar doença em somente um olho.
Embora a mutação genética do gene RB1 aumente o risco de retinoblastoma, isso não significa que a criança vá necessariamente desenvolver o câncer.
Forma não hereditária
Os 70% restantes dos casos de retinoblastoma são da forma não hereditária da doença, que sempre se manifesta como doença unilateral.
Esses pacientes não carregam a mutação do gene RB1 em todas as células do corpo. Acredita-se que a maioria das crianças com doença não hereditária desenvolve um retinoblastoma porque ambas as cópias do gene RB1 são danificadas em uma única célula retiniana em desenvolvimento.
Pacientes que desenvolvem a forma não hereditária não apresentam risco de passar o gene mutante para os seus filhos.
Após o diagnóstico em um membro da família, sugere-se o aconselhamento genético com um especialista, de forma a avaliar a necessidade do exame genético a outros membros da família, especialmente aos irmãos, para determinar o risco de estes desenvolverem a doença.
Sintomas
O retinoblastoma geralmente se apresenta com leucocoria, termo cujo significado literal é “pupila branca”.
Em casos mais óbvios, a leucocoria pode ser identificada por observação simples dos olhos, sem luz especial ou qualquer outra ajuda. Você olha para a pupila da criança e ela tem cor mais clara.
Na maioria dos casos, porém, a pupila parece branca apenas em certas circunstâncias, como quando ela fica dilatada em uma sala com pouca iluminação, quando a criança olha para uma direção específica ou nas fotografias com flash, quando uma das pupilas apresenta um “reflexo branco” anormal em comparação com o outro olho, como demonstrado na foto abaixo.
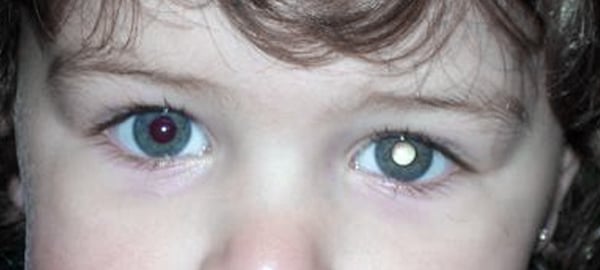
A forma correta de diagnosticar a leucocoria é através do exame dos olhos com um oftalmoscópio. Esse exame pode ser feito facilmente no consultório do pediatra ou de um oftalmologista.
A leucocoria não é uma manifestação exclusiva do retinoblastoma, há outras causas possíveis de pupila branca, como catarata, descolamento de retina, retinopatia da prematuridade, infecção intraocular (endoftalmite) e anormalidades vasculares da retina (doença de Coats).
Além da leucocoria, que é o sinal mais comum, a criança também pode apresentar estrabismo (olhos vesgos), nistagmo (discretos movimentos involuntários dos olhos), olhos vermelhos, dor ocular, inchaço no olho e redução da acuidade visual.
História natural do câncer ocular infantil
Não tratado, o retinoblastoma é uma doença mortal. O tumor cresce dentro do olho e destrói o globo ocular.
A rota mais comum de disseminação metastática é a infiltração direta através do nervo óptico para o sistema nervoso central. Invasão de vasos sanguíneos e disseminação das células tumorais através do sangue para pulmões, ossos, fígado ou cérebro também é comum. Outra via possível é a disseminação através da circulação linfática.
A disseminação metastática geralmente ocorre nos primeiros 12 meses da apresentação clínica do retinoblastoma.
Com tratamento, a taxa de sobrevivência do retinoblastoma é maior que 95%. No entanto, o prognóstico para o salvamento do olho é bem mais baixo e depende do estágio da doença no momento do diagnóstico (ver classificação abaixo).
A regressão espontânea do retinoblastoma já foi descrita em um pequeno número de casos, mas é algo bem raro.
Estágios do retinoblastoma
O retinoblastoma é dividido em dois estágios principais:
Retinoblastoma intraocular
A maioria das crianças é diagnosticada ainda na fase de doença intraocular.
Neste estágio, o câncer ainda está totalmente dentro do olho e não se espalhou. Os tumores nessa fase são classificados em grupos de A a E pelo Sistema de Classificação Internacional para Estadiamento Intraocular, consoante a gravidade da doença.
Grupo A:
- Risco muito baixo de perda do(s) olho(s) afetado(s).
- Tumores pequenos, com 3 milímetros ou menos.
- Os tumores estão somente na retina, a camada de tecido na parte posterior do olho.
- Os tumores não estão próximos da fóvea (o “buraco” central da retina) ou do nervo óptico.
- Nenhum tumor está flutuando no olho, o que é conhecido como semeadura no vítreo. A semeadura do vítreo ocorre quando os tumores se espalham para o vítreo, que é um fluido transparente, semelhante a um gel, que preenche o olho.
- Não há descolamento de retina, uma situação de emergência em que a retina é puxada para fora de sua posição normal (leitura sugerida: Descolamento de retina: sintomas e tratamento).
Grupo B:
- Baixo risco de perda do(s) olho(s) afetado(s).
- Um ou mais tumores são maiores que 3 mm.
- Os tumores estão apenas na retina.
- Não há semeadura no vítreo.
- Não há descolamento de retina a mais de 5 mm da base do tumor
Grupo C:
- Risco moderado de perda do(s) olho(s) afetado(s).
- Os tumores são bem definidos.
- Existe alguma disseminação dos tumores sob a retina, conhecida como semeadura sub-retiniana.
- Os tumores também podem ter se espalhado para o vítreo.
- Há descolamento da retina, com mais de 5 mm da base do tumor até um descolamento completo da retina.
Grupo D:
- Há um alto risco de perda do(s) olho(s) afetado(s).
- A disseminação do tumor é extensa para o vítreo/sob a retina.
- Lesões vítreas ou sub-retinianas do tipo “bolas de neve”.
- Há descolamento de retina, com mais de 5 mm da base do tumor até um descolamento completo da retina.
Grupo E:
- Os retinoblastomas intraoculares classificados como grupo E danificaram extensivamente o olho. Quase não há chance de salvar o(s) olho(s) afetado(s).
- Tumor(es) grande(s) que se estende(m) em direção à parte frontal do olho.
- Glaucoma neovascular. O glaucoma é um dano causado pela pressão dentro do olho. No glaucoma neovascular, essa pressão é causada pelo crescimento de novos vasos sanguíneos na parte frontal do olho como resultado do tumor (leitura sugerida: Glaucoma: sintomas, diagnóstico e tratamento).
- Hemorragia vítrea, que é o sangramento do olho.
- Encolhimento e atrofia do olho.
- Hifema, que é a presença de sangue na parte frontal do olho.
- Intensa vermelhidão e inchaço ao redor dos olhos, semelhante a uma infecção, mas não há infecção de verdade.
Os retinoblastomas intraoculares dos grupos de A a E são curáveis, mas o paciente pode perder olho se tiver a doença nas fases mais avançadas, especialmente D e E.
Retinoblastoma extraocular
O retinoblastoma extraocular é aquele que já se espalhou para além do olho, para os tecidos ao redor dele ou para outras partes do corpo. Esse estágio de tumor é atualmente raro, pois a imensa maioria dos casos é diagnosticada antes.
O retinoblastoma extraocular é bem mais difícil de curar e, se a doença já tiver se espalhado para o sistema nervoso central, a taxa de sobrevivência é menor que 10%.
Diagnóstico
Uma vez que existe a suspeita de retinoblastoma, a criança deve ser encaminhada para um oftalmologista para confirmar — ou descartar — o diagnóstico. Alguns testes utilizados para esse fim são:
- Mapeamento de retina ou exame de fundo de olho: nesse exame, após dilatação da pupila, o médico examina o olho da criança com uma lente especial, tentando identificar o tumor, podendo avaliar o seu tamanho e se há comprometimento de outras estruturas do olho.
- Ultrassonografia ocular: esse exame é fundamental para avaliar o tamanho do tumor, se há descolamento da retina e/ou comprometimento de outras estruturas, como o nervo óptico, por exemplo.
- Tomografia computadorizada ou ressonância magnética: ambos exames podem ajudar no diagnóstico do tumor, mas são mais importantes para avaliar a presença de metástases.
Nas crianças pequenas, menores de 3 anos, pode ser necessário fazer os exames com a criança sedada ou sob anestesia geral.
Rastreio
Nos bebês com elevado risco, isto é, naqueles com história familiar positiva de retinoblastoma, o rastreio do tumor deve ser feito de forma rotineira nos primeiros anos de vida.
A Associação Americana de Oncologistas e Patologistas Oftálmicos sugere o seguinte esquema de rastreio para o retinoblastoma:
- Durante os primeiros três anos de vida, os exames de rastreio são realizados inicialmente a cada um ou dois meses e, em seguida, espaçados a cada três meses, se não houver achados preocupantes.
- Entre os três e sete anos, os exames são realizados a cada quatro a seis meses.
- Se o teste genético revelar que a criança não tem uma mutação herdada do gene RB1, o rastreamento pode ser interrompido aos 7 anos.
- Se o teste genético revelar que a criança é portadora de uma mutação RB1, ela deve continuar a fazer exames regulares a cada um ou dois anos após os sete anos de idade.
Tratamento
Como já referido, o retinoblastoma é uma doença que tem cura. Sendo assim, os objetivos do tratamento são, em ordem de prioridade: curar o tumor e salvar a vida da criança, preservar a visão e preservar a aparência estética do olho. Nem sempre os três objetivos conseguem ser alcançados.
Os melhores tratamentos para o retinoblastoma dependem do tamanho e localização do tumor, se o câncer se espalhou para outras áreas além do olho e da saúde geral da criança.
Algumas opções de tratamento atualmente disponíveis incluem radioterapia, braquiterapia, quimioterapia, crioterapia, fotocoagulação com laser e enucleação (remoção cirúrgica do globo ocular).
Tumores de baixo risco (grupos A ou B) costumam ser tratados por fotocoagulação com laser ou crioterapia. Tumores de moderado risco (grupos C e D) são tratados por quimioterapia, braquiterapia ou radioterapia e laser. Já os tumores graves (alguns do grupo D e grupo E), habitualmente, só podem ser tratados com a remoção completa do globo ocular.
Quando já há invasão do sistema nervoso central ou metástases à distância, o tratamento indicado é a quimioterapia e radioterapia.
Perguntas frequentes
O retinoblastoma tem cura?
Sim, atualmente, a taxa de cura do retinoblastoma é de mais de 95%.
O retinoblastoma pode aparecer em adultos?
Pode, mas o retinoblastoma em pessoas com mais de 7 anos é extremamente raro.
Quais são os sinais e sintomas do retinoblastoma que os pais devem ter atenção?
Os sinais e sintomas típicos do retinoblastoma são:
• Uma pupila que parece branca, em vez do preto normal. Essa alteração é mais perceptível quando uma luz intensa, como um flash de uma câmera fotográfica, incide diretamente nos olhos.
• Olhos desalinhados ou vesgos (estrabismo).
• Problemas de visão.
• Olho vermelho e doloroso.
• Pupila dilatada.
• Íris de cores diferentes (a íris é a parte colorida dos olhos).Qual é o gene relacionado ao retinoblastoma?
Nos pacientes com retinoblastoma hereditário, o tumor se desenvolve como resultado de alterações em um gene específico conhecido como RB1, que está localizado no cromossomo 13 na posição q14.1-q14.2.
O RB1 é o único gene que se sabe estar associado ao retinoblastoma hereditário. A proteína produzida pelo gene RB1 atua como um “supressor de tumor”, o que significa que ela ajuda a evitar que as células cresçam e se dividam muito rapidamente. Se a pessoa herda uma alteração no gene RB1, ela perde essa proteção e fica mais sujeita a ter o retinoblastoma.Quem não tem história familiar de retinoblastoma pode desenvolver a doença?
Sim, é perfeitamente possível e comum uma criança ser o primeiro caso de retinoblastoma na família.
Pacientes com retinoblastoma hereditário têm maior risco de desenvolver outros cânceres?
Sim, pacientes que herdaram uma mutação do gene RB1 também apresentam maior risco de desenvolverem sarcoma osteogênico, sarcomas de tecidos moles (especialmente leiomiossarcoma) e melanoma maligno.
Referências
- Retinoblastoma: Clinical presentation, evaluation, and diagnosis – UpToDate.
- Retinoblastoma: Treatment and outcome – UpToDate.
- Retinoblastoma – Health Professional Version – National Cancer Institute.
- What Is Retinoblastoma? – American Cancer Society.
- Screening Children at Risk for Retinoblastoma: Consensus Report from the American Association of Ophthalmic Oncologists and Pathologists – Ophthalmology.
- Retinoblastoma – Childhood: Stages – American Society of Clinical Oncology (ASCO).
- Yanoff M, et al., eds. Malignant intraocular neoplasms. In: Ophthalmology. 5th ed. Edinburgh, U.K.: Elsevier; 2019.
- Kliegman RM, et al. Retinoblastoma. In: Nelson Textbook of Pediatrics. 20th ed. Philadelphia, Pa.: Elsevier; 2016.
Autor(es)
Médico graduado pela Universidade Federal do Rio de Janeiro (UFRJ), com títulos de especialista em Medicina Interna e Nefrologia pela Universidade Estadual do Rio de Janeiro (UERJ), Sociedade Brasileira de Nefrologia (SBN), Universidade do Porto e pelo Colégio de Especialidade de Nefrologia de Portugal.

O Dr. Renato Oliveira é medico oftalmologista graduado pela Universidade Federal do Rio de Janeiro (UFRJ) com especialização em Córnea e Doenças Externas Oculares pela Universidade Federal de São Paulo (Unifesp). Faz parte do Conselho Brasileiro de Oftalmologia e da Sociedade Brasileira de Catarata e Cirurgia Refrativa