O que é a granulomatose de Wegener?
A granulomatose com poliangiite, antigamente chamada de granulomatose de Wegener, é uma vasculite de origem autoimune, ou seja, uma doença que cursa com uma inflamação difusa dos vasos sanguíneos do corpo, provocada pela produção inapropriada de autoanticorpos contra os próprios vasos sanguíneos.
Essa vasculite acomete todo o corpo, mas ataca principalmente os vasos sanguíneos dos rins, pulmões e vias respiratórias.
O motivo pelo qual ocorre essa desregulação do sistema imune, fazendo com que o nosso corpo comece a se autodestruir, ainda é desconhecido. A inflamação e a destruição dos vasos levam a uma restrição do fluxo sanguíneo para vários tecidos e órgãos.
A granulomatose com poliangiite é uma doença pouco comum, afetando 1 a cada 30.000 pessoas.
A inflamação causada pela doença produz um tipo de tecido inflamatório chamado de granuloma, uma espécie de tumoração microscópica em forma de grânulos. Daí o nome granulomatose de Wegener. Os granulomas destroem os vasos, impedem o aporte de sangue para os órgãos e causam necrose dos tecidos afetados.
Se você não está familiarizado com os conceitos de doença autoimune e vasculite, sugerimos também a leitura dos textos:
Sintomas
A granulomatose de Wegener pode ocorrer em qualquer idade, mas acomete principalmente indivíduos entre os 30 e 50 anos. É mais comum em brancos que em negros e ocorre mais em homens que em mulheres.
A granulomatose com poliangiite é uma doença muito grave, que se não tratada, apresenta uma taxa de mortalidade muito elevada. Antes do advento dos tratamentos mais modernos com imunossupressores, a sobrevida média era de apenas 5 meses, sendo que 90% dos pacientes faleciam com menos de um ano de diagnóstico.
O quadro clínico varia muito de paciente para paciente. Alguns apresentam doença restrita a um único órgão enquanto outros podem cursar com um quadro multissistêmico muito dramático.
A granulomatose de Wegener costuma iniciar-se de forma indolente, com sintomas leves e inespecíficos como cansaço, perda do apetite, febre baixa e desânimo. Os primeiros sintomas específicos costumam ocorrer nas vias aéreas superiores com rinite e sinusite. O quadro pode lembrar o de uma gripe, porém, com a duração dos sintomas muito maior.
Depois de começar de modo brando, a vasculite apresenta um rápido agravamento dos sintomas. A sinusite piora e não responde a nenhum tratamento convencional. A secreção nasal pode se tornar sanguinolenta e deformidades e ulcerações dentro e fora da cavidade nasal podem surgir.
Lesão pulmonar
O acometimento do pulmão na granulomatose com poliangiite é típico. Até 1/3 dos pacientes que cursam com lesão pulmonar apresentam um quadro brando. Os outros 2/3, porém, sofrem com uma doença mais grave.
Os principais sintomas da vasculite pulmonar pelo Wegener são a tosse, a falta de ar e a expectoração com raias de sangue. Em 5% a 10% dos casos, a vasculite das vias respiratórias é muito grave e leva a uma grande hemorragia pulmonar com uma maciça expectoração de sangue. Se não for tratada, a lesão pulmonar pode ser fatal.
Lesão renal
Os rins são outros órgãos frequentemente acometidos pela granulomatose de Wegener. A lesão típica é uma glomerulonefrite necrotizante.
O quadro clínico da glomerulonefrite pelo Wegener se caracteriza por uma insuficiência renal aguda, manifestada por uma rápida e súbita elevação da creatinina associado a hipertensão arterial e hematúria (sangue na urina).
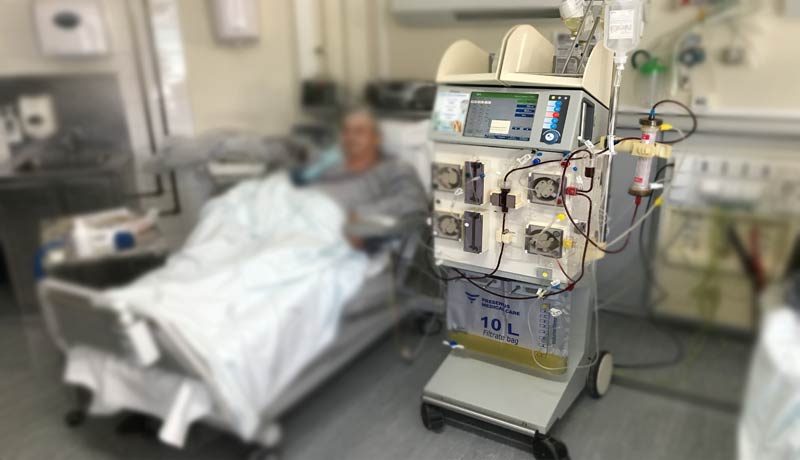
A vasculite renal leva a destruição do tecido funcionante dos rins, e se não tratada a tempo, torna o paciente dependente de hemodiálise.
Não é incomum o acometimento simultâneo dos rins e dos pulmões, chamada de síndrome pulmão-rim. Toda vez que um paciente se apresenta com expectoração sanguinolenta associado a hematúria ou elevação súbita da creatinina, a granulomatose de Wegener é um dos principais diagnósticos diferenciais.
Outras lesões da granulomatose com poliangiite
Além dos sintomas respiratórios e renais, a granulomatose de Wegener pode ainda acometer diversos outros órgãos:
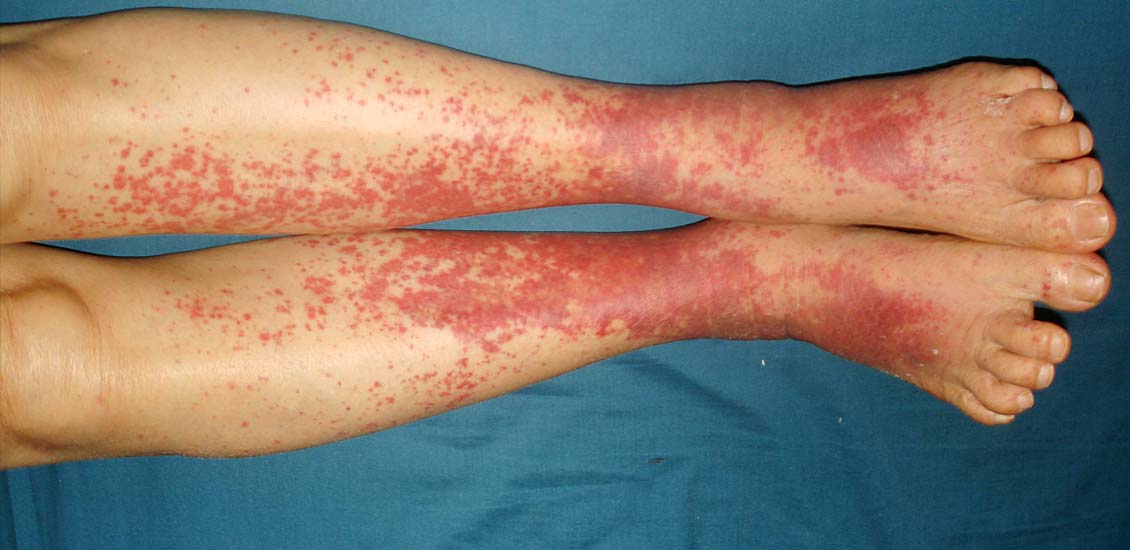
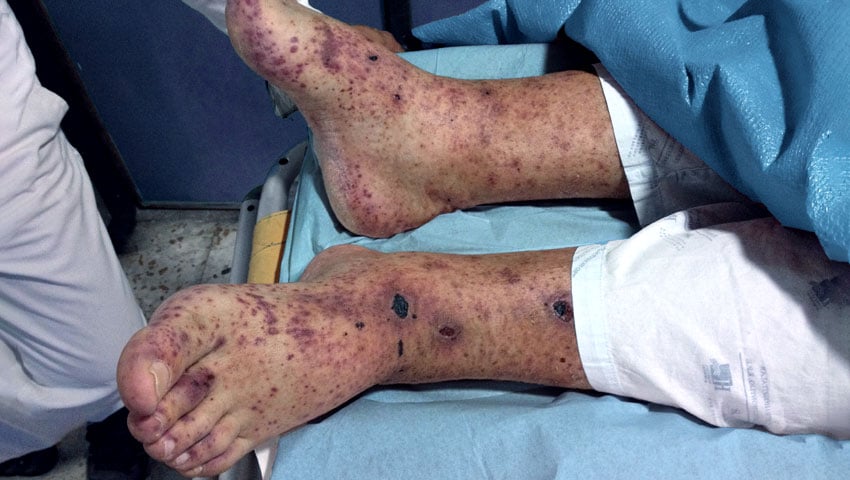
- A vasculite da pele pode se manifestar como bolhas, erupções, úlceras e hemorragias.
- Artrites.
- Lesões neurológicas do sistema nervoso central e dos nervos periféricos.
- O acometimento dos ouvidos também é comum podendo levar a otite e perda da audição.
- Os olhos também podem ser afetados, cursando com conjuntivite, lesão do nervo ocular, oclusão da artéria da retina e outras lesões.
Na verdade, qualquer órgão ou sistema pode ser acometido pela vasculite. O que foi descrito acima são as manifestações mais comuns.
Diagnóstico
Através das análises de sangue é possível detectar um autoanticorpo típico do Wegener, chamado anticorpo anticitoplasma de neutrófilo, ou simplesmente ANCA. A presença deste autoanticorpo é importante para o diagnóstico uma vez que o mesmo está presente em até 90% dos casos, porém, sozinho, não é suficiente.
Para se fechar o diagnóstico do Wegener é preciso uma biópsia, remoção de um pedaço do tecido afetado para avaliação microscópica. Biopsias da nasofaringe ou da pele são mais fáceis de ser realizadas, mas nem sempre há lesão visível para se obter amostras. Muitas vezes é preciso lançar mão de um procedimento mais invasivo para realização das biópsias de pulmão ou dos rins (leia: Biópsia renal).
Tratamento
Como se trata de uma doença de origem imune e potencialmente muito grave, o tratamento é feito à base de imunossupressores fortes. Não existe cura para a granulomatose com poliangiite, porém, a história natural da doença mudou muito nos últimos 20-30 anos.
As principais drogas usadas são os corticoides em doses elevadas e a ciclofosfamida. Outras drogas que também podem ser usadas são a azatioprina, leflunomida, metotrexato ou rituximab.
O uso do antibiótico Bactrim, por motivos ainda não bem esclarecidos, também parece ajudar no controle da doença, além de prevenir infecções oportunistas pela imunossupressão.
Nos casos graves com rápida perda da função renal e hemorragia pulmonar, indica-se a realização da plasmaférese para remoção imediata do excesso de autoanticorpos circulantes.
Com as atuais drogas imunossupressoras, cerca de 75% dos pacientes conseguem atingir remissão completa da doença. Destes, 30% apresentam recaídas ao longo do tempo, necessitando retorno da imunossupressão. Se antigamente mais de 90% dos pacientes faleciam no primeiro ano, hoje em dia, a mortalidade em 5 anos é menor que 20%.
Referências
- Revised 2017 international consensus on testing of ANCAs in granulomatosis with polyangiitis and microscopic polyangiitis – Nature Reviews Rheumatology.
- Small-Vessel Vasculitis – New England Journal of Medicine.
- Granulomatosis with polyangiitis and microscopic polyangiitis: Clinical manifestations and diagnosis – UpToDate.
- Granulomatosis with polyangiitis and microscopic polyangiitis: Initial immunosuppressive therapy – UpToDate.
- Granulomatosis with Polyangiitis – The Vasculitis Foundation.
- Granulomatosis with Polyangiitis (Wegener’s) – American College of Rheumatology.
Autor(es)
Médico graduado pela Universidade Federal do Rio de Janeiro (UFRJ), com títulos de especialista em Medicina Interna e Nefrologia pela Universidade Estadual do Rio de Janeiro (UERJ), Sociedade Brasileira de Nefrologia (SBN), Universidade do Porto e pelo Colégio de Especialidade de Nefrologia de Portugal.