O que é a doença de Fabry?
A doença de Fabry, também chamada de doença de Anderson-Fabry, é uma patologia hereditária rara, transmitida de modo recessivo pelo cromossoma X, o cromossoma sexual feminino.
Estima-se que apenas 1 em cada 20 a 40 mil homens apresente a mutação responsável pelos sintomas da doença.
Os pacientes com doenças de Fabry apresentam uma deficiência de uma enzima chamada alfa-galactosidase A, o que acarreta problemas renais, cardíacos e do sistema nervoso.
Neste artigo explicaremos a doença de Fabry, abordando a sua transmissão genética, as suas causas, os sintomas e as opções de tratamento.
Genética
Todos os humanos possuem dois cromossomas sexuais. Os homens possuem um cromossoma sexual Y e um cromossoma sexual X (homens são XY). Já as mulheres possuem dois cromossomas sexuais X e nenhum cromossoma Y (mulheres são XX).
Cada um dos cromossomas sexuais que possuímos foram transmitidos por um dos pais. Por exemplo, quando um casal tem um filho homem, isso significa que o menino recebeu o cromossoma Y do seu pai e o cromossoma X da mãe, formando, assim, o par XY. Quando o casal tem uma menina, ela recebe um cromossoma X do pai e um cromossoma X da mãe, formando o par XX.
Portanto, como a mãe só é capaz de passar o cromossoma X para os seus filhos, quem “decide” o sexo da criança é sempre o cromossoma de origem paterna. Se o pai passar um Y, será um menino; se ele passar o X, será uma menina.
A doença de Fabry é uma doença transmitida por um cromossoma X defeituoso.
Quando dizemos que o Fabry é uma doença transmitida de modo recessivo pelo cromossoma X, isso significa que para a doença se manifestar nas mulheres elas precisam receber os 2 cromossomas X defeituosos (um do pai e outro da mãe).
Se um cromossoma X for saudável e o outro doente, a mulher não desenvolve a doença, pois o cromossoma saudável a protege desse defeito. Como os homens só apresentam um cromossoma X, eles não têm como ter outro cromossoma X para protegê-lo. Portanto, basta um cromossoma X defeituoso para que ele desenvolva a doença de Fabry.
Pelo que foi explicado até aqui, fica fácil entender por que a doença de Fabry é uma patologia que acomete muito mais os homens que as mulheres. Também é fácil entender por que a doença nos homens é sempre herdada da mãe, já que os humanos do sexo masculino obrigatoriamente recebem o cromossoma Y do pai e cromossoma X da mãe.
Seguindo esse raciocínio, podemos afirmar que os homens que possuem a doença de Fabry não são capazes de transmiti-la a seus filhos homens, dado que esses recebem apenas o seu cromossoma Y. Porém, as filhas de pais com Fabry irão obrigatoriamente receber um cromossoma X defeituoso. Se elas também receberem um cromossoma X doente da mãe, desenvolverão a doença.
Resumindo:
- A doença de Fabry praticamente só ocorre em homens.
- Para uma mulher ter Fabry, ela precisa que ambos os pais tenham o cromossoma X defeituoso*.
- As mulheres costumam ser portadoras assintomáticas do cromossoma X defeituoso, pois o seu outro cromossoma X saudável a protege da doença.
- São as mulheres portadoras cromossoma X defeituoso que transmitem o defeito para os seus filhos.
* Esse conceito tem mudado nos últimos anos, pois há casos reconhecidos de mulheres com apenas um cromossoma X defeituoso que apresentam graus distintos de manifestações clínica da doença de Fabry.
Causas
Como já dito no início do texto, a doença de Fabry acontece por uma deficiência de uma enzima chamada alfa-galactosidase A (alfa-gal A). Essa é uma enzima presente dentro de nossas células que é responsável pela eliminação de uma substância gordurosa chamada globotriaosilceramida (GB3 ou GL-3).
Na ausência da alfa-gal A, a GB3 se acumula dentro das células, depositando-se em vários tecidos, principalmente nas paredes dos vasos sanguíneos. Por isso, a doença de Fabry faz parte de um grupo de patologias chamado de doenças de depósito.
Todos os sinais e sintomas da doença de Fabry são derivados desse acúmulo de GB3, que pode obstruir vasos sanguíneos, causando isquemias e infartos de órgãos e tecidos.
Sintomas
Os sintomas da doença de Fabry costumam seguir um certa ordem de aparecimento. Os primeiros sinais surgem na infância ou início da adolescência e incluem:
Dores nos membros
A dor ocorre principalmente nas mãos e nos pés. É causada por acometimento dos nervos do sistema nervoso periférico. Normalmente, esse é o primeiro sintoma e pode ser desencadeado por estresse, frio, calor ou atividades físicas extenuantes.
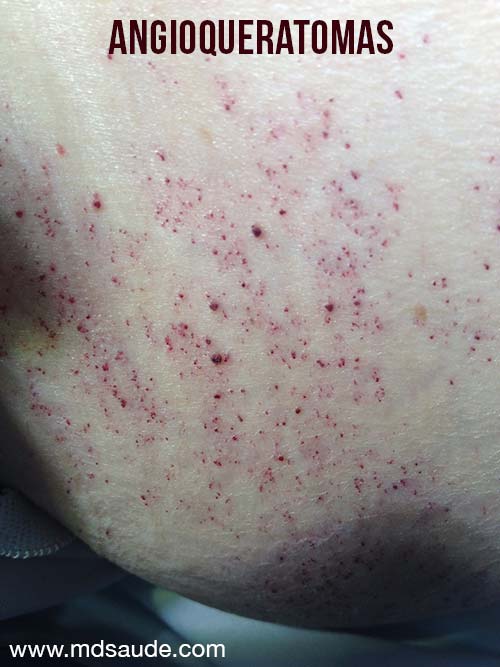
Lesões de pele
Duas lesões são comuns: teleangiectasias e angioqueratomas.
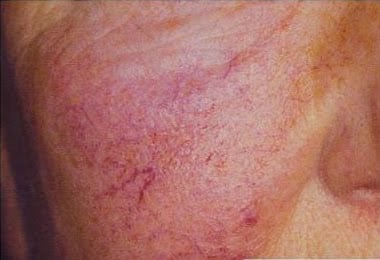
Teleangiectasias são pequenos vasos dilatados na pele, também chamados de aranhas vasculares pelo seu formato. São comumente vistos em pessoas com varizes nas pernas. Já o angioqueratomas são elevações puntiformes arroxadas.
Na doença de Fabry, as duas lesões costumam aparecer juntas, principalmente nas coxas, quadril e em volta do umbigo.
Alterações das glândulas sudoríparas
Outro problema comum no Fabry é o acometimento das glândulas sudoríparas, responsáveis pela produção de suor. Os pacientes podem apresentar baixa taxa de transpiração (hipoidrose), elevada temperatura corporal, fadiga intensa e intolerância ao calor.
Alterações oculares
Depósitos de GB3 nos vasos da córnea dos olhos costumam causar a chamada córnea verticilata, uma lesão da córnea que pode ser detectada com exames oftalmológicos especiais (oftalmoscopia de lâmpada de fenda).
Sintomas gastrointestinais
Sintomas gastrointestinais como dor após alimentação, náuseas e diarreia também são comuns.
Rins
50% dos pacientes com doença de Fabry apresentam acometimento renal. Os primeiros sinais são o aumento da produção de urina por incapacidade do rim em reter água. Depois surgem a proteinúria e, mais adiante, insuficiência renal crônica. Boa parte dos pacientes acaba por necessitar de hemodiálise.
Coração
O envolvimento cardíaco costuma se dar na forma de insuficiência cardíaca, arritmias cardíacas, hipertrofia do ventrículo esquerdo e isquemia do miocárdio.
Cérebro
O acometimento do sistema nervoso central pode se dar através de ataques isquêmicos transitórios ou AVC, tonturas, vertigens ou dor de cabeça.
Doença de Fabry nas mulheres
A maioria das mulheres é portadora de apenas um cromossoma X defeituoso e apresenta poucos ou nenhum sintoma da doença de Fabry.
Porém, por fatores ainda não bem conhecidos, existe um pequeno grupo que desenvolve a doença tal como os homens.
Diagnóstico
Por ser uma doença rara e pouco conhecida, inclusive entre os médicos, muitas vezes os pacientes precisam esperar anos entre os primeiros sintomas e o diagnóstico definitivo. Quando há história familiar, o diagnóstico sai mais facilmente, pois a própria família já sugere a hipótese da doença ao médico.
A tríade de alterações renais + angioqueratomas + dor nos membros em crianças ou jovens do sexo masculino, deve sempre levantar a suspeita de doença de Fabry.
O diagnóstico pode ser confirmado através da dosagem da enzima alfa-galactosidase A. A maioria dos pacientes apresenta níveis indetectáveis ou muito reduzidos.
A biópsia de pele ou do rim pode fornecer o diagnóstico naqueles casos atípicos, no quais o médico não pensa inicialmente na doença de Fabry como diagnóstico diferencial.
As mulheres portadoras do gene da doença podem ser assintomáticas e ter níveis de alfa-gal A normais. Nestes casos apenas testes genéticos podem confirmar se a mesma possui o gene da doença, sendo passível de transmissão para seus filhos. A análise genética ainda é feita em pouquíssimos laboratórios.
Tratamento
A doença de Fabry não tem cura. Porém, já existe uma forma sintética da enzima alfa-gal A para administração. As duas marcas atualmente comercializadas são a Replagal® e a Fabrazyme®.
Recentemente, um novo fármaco foi aprovado na Europa e EUA: Migalastat (nome comercial Galafold).
O Migalastat liga-se a determinadas formas instáveis da alfa-gal A, estabilizando a enzima. Isto permite que a enzima seja transportada para áreas da célula onde pode decompor a GL-3.
Atualmente, a expectativa de vida média dos pacientes não tratados está ao redor dos 50 anos. Os medicamentos são muito recentes e ainda não houve tempo para se comprovar diminuição da mortalidade, apesar das evidências de existir redução dos depósitos de GB3 e retardamento da perda de função renal.
Quanto mais cedo for iniciado o medicamento, melhores são os resultados. O tratamento é para vida toda.
Referências
- Fabry Disease – National Organization for Rare Disorders.
- Fabry disease – U.S National Library of Medicine.
- Fabry disease: Clinical features and diagnosis – UpToDate.
- Fabry disease: Treatment – UpToDate.
- Fabry Disease – Medscape.
Autor(es)
Médico graduado pela Universidade Federal do Rio de Janeiro (UFRJ), com títulos de especialista em Medicina Interna e Nefrologia pela Universidade Estadual do Rio de Janeiro (UERJ), Sociedade Brasileira de Nefrologia (SBN), Universidade do Porto e pelo Colégio de Especialidade de Nefrologia de Portugal.